This is an old revision of the document!
XAS $L_{2,3}$
Once the ground-state is calculated one can calculate the spectra. This example shows the Ni $2p$ to $3d$ excitations in NiO. Note that these excitations have an energy of more than 800 electron Volt, which is much higher than the chemically relevant energy scales. Non-the-less these kind of spectroscopy contain useful information on the local ground-state wave-function and the low energy effective Hamiltonian.
This tutorial compares calculated spectra to experiment. In order to make the plots you need to download the experimental data. You can download them in a zip file here nio_data.zip. Please unpack this file and make sure to have the folders NiO_Experiment and NiO_Radial in the same folder as you do the calculations. And as always, if used in a publication, please cite the original papers that published the data.
The input file to do these calculation is:
- XAS.Quanty
Verbosity(0) -- here we calculate the 2p to 3d x-ray absorption of NiO within the Ligand-field theory -- approximation. The first part of the script is very much the same as calculating -- the ground-state with the addition that we now also need a 2p core shell in the basis -- from the previous example we know that within NiO there are 3 states close to each other -- and then there is an energy gap of about 1 eV. We thus only need to consider the 3 -- lowest states (Npsi=3 later on) NF=26 NB=0 IndexDn_2p={ 0, 2, 4} IndexUp_2p={ 1, 3, 5} IndexDn_3d={ 6, 8,10,12,14} IndexUp_3d={ 7, 9,11,13,15} IndexDn_Ld={16,18,20,22,24} IndexUp_Ld={17,19,21,23,25} -- angular momentum operators on the d-shell OppSx_3d =NewOperator("Sx" ,NF, IndexUp_3d, IndexDn_3d) OppSy_3d =NewOperator("Sy" ,NF, IndexUp_3d, IndexDn_3d) OppSz_3d =NewOperator("Sz" ,NF, IndexUp_3d, IndexDn_3d) OppSsqr_3d =NewOperator("Ssqr" ,NF, IndexUp_3d, IndexDn_3d) OppSplus_3d=NewOperator("Splus",NF, IndexUp_3d, IndexDn_3d) OppSmin_3d =NewOperator("Smin" ,NF, IndexUp_3d, IndexDn_3d) OppLx_3d =NewOperator("Lx" ,NF, IndexUp_3d, IndexDn_3d) OppLy_3d =NewOperator("Ly" ,NF, IndexUp_3d, IndexDn_3d) OppLz_3d =NewOperator("Lz" ,NF, IndexUp_3d, IndexDn_3d) OppLsqr_3d =NewOperator("Lsqr" ,NF, IndexUp_3d, IndexDn_3d) OppLplus_3d=NewOperator("Lplus",NF, IndexUp_3d, IndexDn_3d) OppLmin_3d =NewOperator("Lmin" ,NF, IndexUp_3d, IndexDn_3d) OppJx_3d =NewOperator("Jx" ,NF, IndexUp_3d, IndexDn_3d) OppJy_3d =NewOperator("Jy" ,NF, IndexUp_3d, IndexDn_3d) OppJz_3d =NewOperator("Jz" ,NF, IndexUp_3d, IndexDn_3d) OppJsqr_3d =NewOperator("Jsqr" ,NF, IndexUp_3d, IndexDn_3d) OppJplus_3d=NewOperator("Jplus",NF, IndexUp_3d, IndexDn_3d) OppJmin_3d =NewOperator("Jmin" ,NF, IndexUp_3d, IndexDn_3d) Oppldots_3d=NewOperator("ldots",NF, IndexUp_3d, IndexDn_3d) -- Angular momentum operators on the Ligand shell OppSx_Ld =NewOperator("Sx" ,NF, IndexUp_Ld, IndexDn_Ld) OppSy_Ld =NewOperator("Sy" ,NF, IndexUp_Ld, IndexDn_Ld) OppSz_Ld =NewOperator("Sz" ,NF, IndexUp_Ld, IndexDn_Ld) OppSsqr_Ld =NewOperator("Ssqr" ,NF, IndexUp_Ld, IndexDn_Ld) OppSplus_Ld=NewOperator("Splus",NF, IndexUp_Ld, IndexDn_Ld) OppSmin_Ld =NewOperator("Smin" ,NF, IndexUp_Ld, IndexDn_Ld) OppLx_Ld =NewOperator("Lx" ,NF, IndexUp_Ld, IndexDn_Ld) OppLy_Ld =NewOperator("Ly" ,NF, IndexUp_Ld, IndexDn_Ld) OppLz_Ld =NewOperator("Lz" ,NF, IndexUp_Ld, IndexDn_Ld) OppLsqr_Ld =NewOperator("Lsqr" ,NF, IndexUp_Ld, IndexDn_Ld) OppLplus_Ld=NewOperator("Lplus",NF, IndexUp_Ld, IndexDn_Ld) OppLmin_Ld =NewOperator("Lmin" ,NF, IndexUp_Ld, IndexDn_Ld) OppJx_Ld =NewOperator("Jx" ,NF, IndexUp_Ld, IndexDn_Ld) OppJy_Ld =NewOperator("Jy" ,NF, IndexUp_Ld, IndexDn_Ld) OppJz_Ld =NewOperator("Jz" ,NF, IndexUp_Ld, IndexDn_Ld) OppJsqr_Ld =NewOperator("Jsqr" ,NF, IndexUp_Ld, IndexDn_Ld) OppJplus_Ld=NewOperator("Jplus",NF, IndexUp_Ld, IndexDn_Ld) OppJmin_Ld =NewOperator("Jmin" ,NF, IndexUp_Ld, IndexDn_Ld) -- total angular momentum OppSx = OppSx_3d + OppSx_Ld OppSy = OppSy_3d + OppSy_Ld OppSz = OppSz_3d + OppSz_Ld OppSsqr = OppSx * OppSx + OppSy * OppSy + OppSz * OppSz OppLx = OppLx_3d + OppLx_Ld OppLy = OppLy_3d + OppLy_Ld OppLz = OppLz_3d + OppLz_Ld OppLsqr = OppLx * OppLx + OppLy * OppLy + OppLz * OppLz OppJx = OppJx_3d + OppJx_Ld OppJy = OppJy_3d + OppJy_Ld OppJz = OppJz_3d + OppJz_Ld OppJsqr = OppJx * OppJx + OppJy * OppJy + OppJz * OppJz -- define the coulomb operator -- we here define the part depending on F0 seperately from the part depending on F2 -- when summing we can put in the numerical values of the slater integrals OppF0_3d =NewOperator("U", NF, IndexUp_3d, IndexDn_3d, {1,0,0}) OppF2_3d =NewOperator("U", NF, IndexUp_3d, IndexDn_3d, {0,1,0}) OppF4_3d =NewOperator("U", NF, IndexUp_3d, IndexDn_3d, {0,0,1}) -- define onsite energies - crystal field -- Akm = {{k1,m1,Akm1},{k2,m2,Akm2}, ... } Akm = PotentialExpandedOnClm("Oh", 2, {0.6,-0.4}) OpptenDq_3d = NewOperator("CF", NF, IndexUp_3d, IndexDn_3d, Akm) OpptenDq_Ld = NewOperator("CF", NF, IndexUp_Ld, IndexDn_Ld, Akm) Akm = PotentialExpandedOnClm("Oh", 2, {1,0}) OppNeg_3d = NewOperator("CF", NF, IndexUp_3d, IndexDn_3d, Akm) OppNeg_Ld = NewOperator("CF", NF, IndexUp_Ld, IndexDn_Ld, Akm) Akm = PotentialExpandedOnClm("Oh", 2, {0,1}) OppNt2g_3d = NewOperator("CF", NF, IndexUp_3d, IndexDn_3d, Akm) OppNt2g_Ld = NewOperator("CF", NF, IndexUp_Ld, IndexDn_Ld, Akm) OppNUp_2p = NewOperator("Number", NF, IndexUp_2p, IndexUp_2p, {1,1,1}) OppNDn_2p = NewOperator("Number", NF, IndexDn_2p, IndexDn_2p, {1,1,1}) OppN_2p = OppNUp_2p + OppNDn_2p OppNUp_3d = NewOperator("Number", NF, IndexUp_3d, IndexUp_3d, {1,1,1,1,1}) OppNDn_3d = NewOperator("Number", NF, IndexDn_3d, IndexDn_3d, {1,1,1,1,1}) OppN_3d = OppNUp_3d + OppNDn_3d OppNUp_Ld = NewOperator("Number", NF, IndexUp_Ld, IndexUp_Ld, {1,1,1,1,1}) OppNDn_Ld = NewOperator("Number", NF, IndexDn_Ld, IndexDn_Ld, {1,1,1,1,1}) OppN_Ld = OppNUp_Ld + OppNDn_Ld -- define L-d interaction Akm = PotentialExpandedOnClm("Oh", 2, {1,0}) OppVeg = NewOperator("CF", NF, IndexUp_3d, IndexDn_3d, IndexUp_Ld, IndexDn_Ld,Akm) + NewOperator("CF", NF, IndexUp_Ld, IndexDn_Ld, IndexUp_3d, IndexDn_3d, Akm) Akm = PotentialExpandedOnClm("Oh", 2, {0,1}) OppVt2g = NewOperator("CF", NF, IndexUp_3d, IndexDn_3d, IndexUp_Ld, IndexDn_Ld,Akm) + NewOperator("CF", NF, IndexUp_Ld, IndexDn_Ld, IndexUp_3d, IndexDn_3d, Akm) -- core valence interaction Oppcldots= NewOperator("ldots", NF, IndexUp_2p, IndexDn_2p) OppUpdF0 = NewOperator("U", NF, IndexUp_2p, IndexDn_2p, IndexUp_3d, IndexDn_3d, {1,0}, {0,0}) OppUpdF2 = NewOperator("U", NF, IndexUp_2p, IndexDn_2p, IndexUp_3d, IndexDn_3d, {0,1}, {0,0}) OppUpdG1 = NewOperator("U", NF, IndexUp_2p, IndexDn_2p, IndexUp_3d, IndexDn_3d, {0,0}, {1,0}) OppUpdG3 = NewOperator("U", NF, IndexUp_2p, IndexDn_2p, IndexUp_3d, IndexDn_3d, {0,0}, {0,1}) -- dipole transition t=math.sqrt(1/2) Akm = {{1,-1,t},{1, 1,-t}} TXASx = NewOperator("CF", NF, IndexUp_3d, IndexDn_3d, IndexUp_2p, IndexDn_2p, Akm) Akm = {{1,-1,t*I},{1, 1,t*I}} TXASy = NewOperator("CF", NF, IndexUp_3d, IndexDn_3d, IndexUp_2p, IndexDn_2p, Akm) Akm = {{1,0,1}} TXASz = NewOperator("CF", NF, IndexUp_3d, IndexDn_3d, IndexUp_2p, IndexDn_2p, Akm) TXASr = t*(TXASx - I * TXASy) TXASl =-t*(TXASx + I * TXASy) -- We follow the energy definitions as introduced in the group of G.A. Sawatzky (Groningen) -- J. Zaanen, G.A. Sawatzky, and J.W. Allen PRL 55, 418 (1985) -- for parameters of specific materials see -- A.E. Bockquet et al. PRB 55, 1161 (1996) -- After some initial discussion the energies U and Delta refer to the center of a configuration -- The L^10 d^n configuration has an energy 0 -- The L^9 d^n+1 configuration has an energy Delta -- The L^8 d^n+2 configuration has an energy 2*Delta+Udd -- -- If we relate this to the onsite energy of the L and d orbitals we find -- 10 eL + n ed + n(n-1) U/2 == 0 -- 9 eL + (n+1) ed + (n+1)n U/2 == Delta -- 8 eL + (n+2) ed + (n+1)(n+2) U/2 == 2*Delta+U -- 3 equations with 2 unknowns, but with interdependence yield: -- ed = (10*Delta-nd*(19+nd)*U/2)/(10+nd) -- eL = nd*((1+nd)*Udd/2-Delta)/(10+nd) -- -- For the final state we/they defined -- The 2p^5 L^10 d^n+1 configuration has an energy 0 -- The 2p^5 L^9 d^n+2 configuration has an energy Delta + Udd - Upd -- The 2p^5 L^8 d^n+3 configuration has an energy 2*Delta + 3*Udd - 2*Upd -- -- If we relate this to the onsite energy of the p and d orbitals we find -- 6 ep + 10 eL + n ed + n(n-1) Udd/2 + 6 n Upd == 0 -- 6 ep + 9 eL + (n+1) ed + (n+1)n Udd/2 + 6 (n+1) Upd == Delta -- 6 ep + 8 eL + (n+2) ed + (n+1)(n+2) Udd/2 + 6 (n+2) Upd == 2*Delta+Udd -- 5 ep + 10 eL + (n+1) ed + (n+1)(n) Udd/2 + 5 (n+1) Upd == 0 -- 5 ep + 9 eL + (n+2) ed + (n+2)(n+1) Udd/2 + 5 (n+2) Upd == Delta+Udd-Upd -- 5 ep + 8 eL + (n+3) ed + (n+3)(n+2) Udd/2 + 5 (n+3) Upd == 2*Delta+3*Udd-2*Upd -- 6 equations with 3 unknowns, but with interdependence yield: -- epfinal = (10*Delta + (1+nd)*(nd*Udd/2-(10+nd)*Upd) / (16+nd) -- edfinal = (10*Delta - nd*(31+nd)*Udd/2-90*Upd) / (16+nd) -- eLfinal = ((1+nd)*(nd*Udd/2+6*Upd)-(6+nd)*Delta) / (16+nd) -- -- -- -- note that ed-ep = Delta - nd * U and not Delta -- note furthermore that ep and ed here are defined for the onsite energy if the system had -- locally nd electrons in the d-shell. In DFT or Hartree Fock the d occupation is in the end not -- nd and thus the onsite energy of the Kohn-Sham orbitals is not equal to ep and ed in model -- calculations. -- -- note furthermore that ep and eL actually should be different for most systems. We happily ignore this fact -- -- We normally take U and Delta as experimentally determined parameters -- number of electrons (formal valence) nd = 8 -- parameters from experiment (core level PES) Udd = 7.3 Upd = 8.5 Delta = 4.7 -- parameters obtained from DFT (PRB 85, 165113 (2012)) F2dd = 11.14 F4dd = 6.87 F2pd = 6.67 G1pd = 4.92 G3pd = 2.80 tenDq = 0.56 tenDqL = 1.44 Veg = 2.06 Vt2g = 1.21 zeta_3d = 0.081 zeta_2p = 11.51 Bz = 0.000001 Hz = 0.120 ed = (10*Delta-nd*(19+nd)*Udd/2)/(10+nd) eL = nd*((1+nd)*Udd/2-Delta)/(10+nd) epfinal = (10*Delta + (1+nd)*(nd*Udd/2-(10+nd)*Upd)) / (16+nd) edfinal = (10*Delta - nd*(31+nd)*Udd/2-90*Upd) / (16+nd) eLfinal = ((1+nd)*(nd*Udd/2+6*Upd) - (6+nd)*Delta) / (16+nd) F0dd = Udd + (F2dd+F4dd) * 2/63 F0pd = Upd + (1/15)*G1pd + (3/70)*G3pd Hamiltonian = F0dd*OppF0_3d + F2dd*OppF2_3d + F4dd*OppF4_3d + zeta_3d*Oppldots_3d + Bz*(2*OppSz_3d + OppLz_3d) + Hz * OppSz_3d + tenDq*OpptenDq_3d + tenDqL*OpptenDq_Ld + Veg * OppVeg + Vt2g * OppVt2g + ed * OppN_3d + eL * OppN_Ld XASHamiltonian = F0dd*OppF0_3d + F2dd*OppF2_3d + F4dd*OppF4_3d + zeta_3d*Oppldots_3d + Bz*(2*OppSz_3d + OppLz_3d)+ Hz * OppSz_3d + tenDq*OpptenDq_3d + tenDqL*OpptenDq_Ld + Veg * OppVeg + Vt2g * OppVt2g + edfinal * OppN_3d + eLfinal * OppN_Ld + epfinal * OppN_2p + zeta_2p * Oppcldots + F0pd * OppUpdF0 + F2pd * OppUpdF2 + G1pd * OppUpdG1 + G3pd * OppUpdG3 -- we now can create the lowest Npsi eigenstates: Npsi=3 -- in order to make sure we have a filling of 8 electrons we need to define some restrictions StartRestrictions = {NF, NB, {"000000 1111111111 0000000000",8,8}, {"111111 0000000000 1111111111",16,16}} psiList = Eigensystem(Hamiltonian, StartRestrictions, Npsi) oppList={Hamiltonian, OppSsqr, OppLsqr, OppJsqr, OppSz_3d, OppLz_3d, Oppldots_3d, OppF2_3d, OppF4_3d, OppNeg_3d, OppNt2g_3d, OppNeg_Ld, OppNt2g_Ld, OppN_3d} -- print of some expectation values print(" # <E> <S^2> <L^2> <J^2> <S_z^3d> <L_z^3d> <l.s> <F[2]> <F[4]> <Neg^3d> <Nt2g^3d><Neg^Ld> <Nt2g^Ld><N^3d>"); for i = 1,#psiList do io.write(string.format("%3i ",i)) for j = 1,#oppList do expectationvalue = Chop(psiList[i]*oppList[j]*psiList[i]) io.write(string.format("%8.3f ",expectationvalue)) end io.write("\n") end -- We calculate the x-ray absorption spectra for z, right circular and left circular polarized light for the 3 lowest eigen-states. (9 spectra in total) XASSpectra = CreateSpectra(XASHamiltonian, {TXASz, TXASr, TXASl}, psiList, {{"Emin",-15}, {"Emax",25}, {"NE",2000}, {"Gamma",0.1}}) -- and put some additional energy broadening on it (0.4 Gaussian and energy dependent lorenzian) XASSpectra.Broaden(0.4, {{-3.7, 0.45}, {-2.2, 0.65}, { 0.0, 0.65}, { 1.0, 2.00}, { 6 , 2.00}, { 8 , 0.80}, {13.2, 0.80}, {14.0, 0.90}, {16.0, 0.90}, {17.0, 2.00}}) -- we can use several operations on a spectrum object. For one we can sum several spectra -- the function Spectra.Sum sums the spectra with the prefactors given in the list afterwards -- sum the spectra of the ground-state for different polarizations in order to get the -- isotropic spectrum XASIsoSpectra = Spectra.Sum(XASSpectra,{1,0,0, 1,0,0, 1,0,0}) -- We can print the spectra to file XASSpectra.Print({{"file","XASSpec.dat"}}) XASIsoSpectra.Print({{"file","XASIsoSpec.dat"}}) -- In order to make plots I use Gnu-plot, which can be called from within Quanty (if it is installed on your computer) -- Here a script to make the plots gnuplotInput = [[ set autoscale set xtic auto set ytic auto set style line 1 lt 1 lw 1 lc rgb "#000000" set style line 2 lt 1 lw 1 lc rgb "#FF0000" set style line 3 lt 1 lw 3 lc rgb "#000000" set xlabel "E (eV)" font "Times,12" set ylabel "Intensity (arb. units)" font "Times,12" set out 'XASSpec.ps' set size 1.0, 1.0 set terminal postscript portrait enhanced color "Times" 12 set multiplot layout 3, 3 plot "XASSpec.dat" u 1:(-$3 ) title 'z-polarized Sz=-1' with lines ls 1 plot "XASSpec.dat" u 1:(-$5 ) title 'z-polarized Sz= 0' with lines ls 1 plot "XASSpec.dat" u 1:(-$7 ) title 'z-polarized Sz= 1' with lines ls 1 plot "XASSpec.dat" u 1:(-$9 ) title 'r-polarized Sz=-1' with lines ls 1 plot "XASSpec.dat" u 1:(-$11) title 'r-polarized Sz= 0' with lines ls 1 plot "XASSpec.dat" u 1:(-$13) title 'r-polarized Sz= 1' with lines ls 1 plot "XASSpec.dat" u 1:(-$15) title 'l-polarized Sz=-1' with lines ls 1 plot "XASSpec.dat" u 1:(-$17) title 'l-polarized Sz= 0' with lines ls 1 plot "XASSpec.dat" u 1:(-$19) title 'l-polarized Sz= 1' with lines ls 1 unset multiplot energyshift=857.6 intensityscale=64 plot "XASSpec.dat" using ($1+energyshift):((-$3-$9-$15) * intensityscale) title 'isotropic theory' with lines ls 1,\ "NiO_Experiment/XAS_L23_PRB_57_11623_1998" using 1:2 title 'isotropic experiment' with lines ls 2 set size 1.0, 0.6 intensityscale=48 set out 'XASIsoSpec.ps' set xrange [847:877] plot "NiO_Experiment/XAS_L23_PRB_57_11623_1998" using 1:2 title 'isotropic experiment' with filledcurves y1=0,\ "XASIsoSpec.dat" using ($1+energyshift):((-$3) * intensityscale) title 'isotropic theory' with lines ls 3 ]] -- write the gnuplot script to a file file = io.open("XASSpec.gnuplot", "w") file:write(gnuplotInput) file:close() -- and finally call gnuplot to execute the script os.execute("gnuplot XASSpec.gnuplot") -- as I like pdf to view and eps to include in the manuel I transform the format os.execute(" ps2pdf XASSpec.ps ; ps2pdf XASIsoSpec.ps ; ps2eps XASIsoSpec.ps ; mv XASIsoSpec.eps temp.eps ; eps2eps temp.eps XASIsoSpec.eps ; rm temp.eps")
The resulting spectra are: (Experimental data from Alders \textit{et al.} \cite{Alders:1995uu}.)
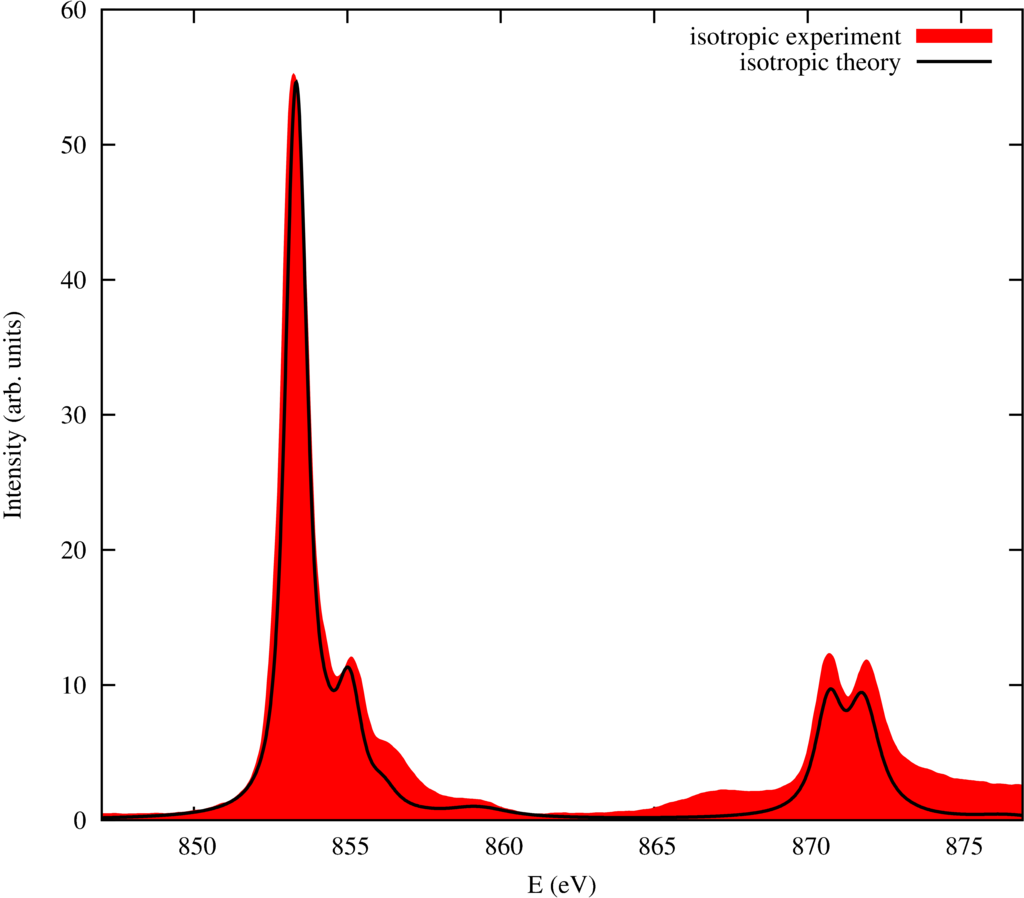 |
| Isotropic spectrum of NiO black theory, red experiment |
|---|
For NiO these spectra already are quite well reproduced on a crystal-field level. This reflects the strongly insulator and ionic behavior of NiO. There are still differences though in the energy range between 845 and 860 eV. In the next section we will calculate these spectra on a ligand field theory level and thereby improving the agrement between theory and experiment substantially.
The output of the script is:
- XAS.out
# <E> <S^2> <L^2> <J^2> <S_z^3d> <L_z^3d> <l.s> <F[2]> <F[4]> <Neg^3d> <Nt2g^3d><Neg^Ld> <Nt2g^Ld><N^3d> 1 -3.503 1.999 12.000 15.095 -0.908 -0.281 -0.305 -1.042 -0.924 2.186 5.990 3.825 6.000 8.175 2 -3.395 1.999 12.000 15.160 -0.004 -0.002 -0.322 -1.043 -0.925 2.189 5.988 3.823 6.000 8.178 3 -3.286 1.999 12.000 15.211 0.903 0.278 -0.336 -1.043 -0.925 2.193 5.987 3.820 6.000 8.180