nIXS $M_{4,5}$
Inelastic x-ray scattering IXS (non-resonant) nIXS or x-ray Raman scattering allows one to measure non-dipolar allowed transitions. A powerful technique to look at even $d$-$d$ transitions with well defined selection rules \cite{Haverkort:2007bv, vanVeenendaal:2008kv, Hiraoka:2011cq}, but can also be used to determine orbital occupations of rare-earth ions that are fundamentally not possible to determine using dipolar spectroscopy \cite{Willers:2012bz}.
This tutorial compares calculated spectra to experiment. In order to make the plots you need to download the experimental data. You can download them in a zip file here nio_data.zip. Please unpack this file and make sure to have the folders NiO_Experiment and NiO_Radial in the same folder as you do the calculations. And as always, if used in a publication, please cite the original papers that published the data.
The first example shows low energy $d$-$d$ transitions in NiO. The input is:
- NIXS_dd.Quanty
-- This example calculates the d-d excitations in NiO using non-resonant Inelastic X-ray -- Scattering. This is one of the most beautiful spectroscopy techniques as the selection -- rules are very "simple" and straight forward. -- We use the A^2 term of the interaction to make transitions between states with photons -- of much higher energy. These photons now cary non negligible momentum and one can make -- transitions beyond the dipole limit. -- Here we look at k=2 and k=4 transitions between the Ni 3d orbitals -- We use the definitions of all operators and basis orbitals as defined in the file -- include and can afterwards directly continue by creating the Hamiltonian -- and calculating the spectra dofile("Include.Quanty") -- The parameters and scheme needed only includes the ground-state (d^8) configuration -- We follow the energy definitions as introduced in the group of G.A. Sawatzky (Groningen) -- J. Zaanen, G.A. Sawatzky, and J.W. Allen PRL 55, 418 (1985) -- for parameters of specific materials see -- A.E. Bockquet et al. PRB 55, 1161 (1996) -- After some initial discussion the energies U and Delta refer to the center of a configuration -- The L^10 d^n configuration has an energy 0 -- The L^9 d^n+1 configuration has an energy Delta -- The L^8 d^n+2 configuration has an energy 2*Delta+Udd -- -- If we relate this to the onsite energy of the L and d orbitals we find -- 10 eL + n ed + n(n-1) U/2 == 0 -- 9 eL + (n+1) ed + (n+1)n U/2 == Delta -- 8 eL + (n+2) ed + (n+1)(n+2) U/2 == 2*Delta+U -- 3 equations with 2 unknowns, but with interdependence yield: -- ed = (10*Delta-nd*(19+nd)*U/2)/(10+nd) -- eL = nd*((1+nd)*Udd/2-Delta)/(10+nd) -- -- note that ed-ep = Delta - nd * U and not Delta -- note furthermore that ep and ed here are defined for the onsite energy if the system had -- locally nd electrons in the d-shell. In DFT or Hartree Fock the d occupation is in the end not -- nd and thus the onsite energy of the Kohn-Sham orbitals is not equal to ep and ed in model -- calculations. -- -- note furthermore that ep and eL actually should be different for most systems. We happily ignore this fact -- -- We normally take U and Delta as experimentally determined parameters -- number of electrons (formal valence) nd = 8 -- parameters from experiment (core level PES) Udd = 7.3 Delta = 4.7 -- parameters obtained from DFT (PRB 85, 165113 (2012)) F2dd = 11.14 F4dd = 6.87 F2pd = 6.67 tenDq = 0.56 tenDqL = 1.44 Veg = 2.06 Vt2g = 1.21 zeta_3d = 0.081 Bz = 0.000001 H112 = 0.120 ed = (10*Delta-nd*(19+nd)*Udd/2)/(10+nd) eL = nd*((1+nd)*Udd/2-Delta)/(10+nd) F0dd = Udd + (F2dd+F4dd) * 2/63 Hamiltonian = F0dd*OppF0_3d + F2dd*OppF2_3d + F4dd*OppF4_3d + zeta_3d*Oppldots_3d + Bz*(2*OppSz_3d + OppLz_3d) + H112 * (OppSx_3d+OppSy_3d+2*OppSz_3d)/sqrt(6) + tenDq*OpptenDq_3d + tenDqL*OpptenDq_Ld + Veg * OppVeg + Vt2g * OppVt2g + ed * OppN_3d + eL * OppN_Ld -- we now can create the lowest Npsi eigenstates: Npsi=3 -- in order to make sure we have a filling of 8 electrons we need to define some restrictions StartRestrictions = {NF, NB, {"000000 00 1111111111 0000000000",8,8}, {"111111 11 0000000000 1111111111",18,18}} psiList = Eigensystem(Hamiltonian, StartRestrictions, Npsi) oppList={Hamiltonian, OppSsqr, OppLsqr, OppJsqr, OppSx_3d, OppLx_3d, OppSy_3d, OppLy_3d, OppSz_3d, OppLz_3d, Oppldots_3d, OppF2_3d, OppF4_3d, OppNeg_3d, OppNt2g_3d, OppNeg_Ld, OppNt2g_Ld, OppN_3d} -- print of some expectation values print(" # <E> <S^2> <L^2> <J^2> <S_x^3d> <L_x^3d> <S_y^3d> <L_y^3d> <S_z^3d> <L_z^3d> <l.s> <F[2]> <F[4]> <Neg^3d> <Nt2g^3d><Neg^Ld> <Nt2g^Ld><N^3d>"); for i = 1,#psiList do io.write(string.format("%3i ",i)) for j = 1,#oppList do expectationvalue = Chop(psiList[i]*oppList[j]*psiList[i]) io.write(string.format("%8.3f ",expectationvalue)) end io.write("\n") end -- in order to calculate nIXS we need to determine the intensity ratio for the different multipole intensities -- ( see PRL 99, 257401 (2007) for the formalism ) -- in short the A^2 interaction is expanded on spherical harmonics and Bessel functions -- The 3d Wannier functions are expanded on spherical harmonics and a radial wave function -- For the radial wave-function we calculate <R(r) | j_k(q r) | R(r)> -- which defines the transition strength for the multipole of order k -- The radial functions here are calculated for a Ni 2+ atom and stored in the folder NiO_Radial -- more sophisticated methods can be used -- read the radial wave functions -- order of functions -- r 1S 2S 2P 3S 3P 3D file = io.open( "NiO_Radial/RnlNi_Atomic_Hartree_Fock", "r") Rnl = {} for line in file:lines() do RnlLine={} for i in string.gmatch(line, "%S+") do table.insert(RnlLine,i) end table.insert(Rnl,RnlLine) end -- some constants a0 = 0.52917721092 Rydberg = 13.60569253 Hartree = 2*Rydberg -- dd transitions from 3d (index 7 in Rnl) to 3d (index 7 in Rnl) -- <R(r) | j_k(q r) | R(r)> function RjRdd (q) Rj0R = 0 Rj2R = 0 Rj4R = 0 dr = Rnl[3][1]-Rnl[2][1] r0 = Rnl[2][1]-2*dr for ir = 2, #Rnl, 1 do r = r0 + ir * dr Rj0R = Rj0R + Rnl[ir][7] * SphericalBesselJ(0,q*r) * Rnl[ir][7] * dr Rj2R = Rj2R + Rnl[ir][7] * SphericalBesselJ(2,q*r) * Rnl[ir][7] * dr Rj4R = Rj4R + Rnl[ir][7] * SphericalBesselJ(4,q*r) * Rnl[ir][7] * dr end return Rj0R, Rj2R, Rj4R end -- the angular part is given as C(theta_q, phi_q)^* C(theta_r, phi_r) -- which is a potential expanded on spherical harmonics function ExpandOnClm(k,theta,phi,scale) ret={} for m=-k, k, 1 do table.insert(ret,{k,m,scale * SphericalHarmonicC(k,m,theta,phi)}) end return ret end -- define nIXS transition operators function TnIXS_dd(q, theta, phi) Rj0R, Rj2R, Rj4R = RjRdd(q) k=0 A0 = ExpandOnClm(k, theta, phi, I^k*(2*k+1)*Rj0R) T0 = NewOperator("CF", NF, IndexUp_3d, IndexDn_3d, A0) k=2 A2 = ExpandOnClm(k, theta, phi, I^k*(2*k+1)*Rj2R) T2 = NewOperator("CF", NF, IndexUp_3d, IndexDn_3d, A2) k=4 A4 = ExpandOnClm(k, theta, phi, I^k*(2*k+1)*Rj4R) T4 = NewOperator("CF", NF, IndexUp_3d, IndexDn_3d, A4) T = T0+T2+T4 T.Chop() return T end -- q in units per a0 (if you want in units per A take 5*a0 to have a q of 5 per A) q=4.5 print("for q=",q," per a0 (",q / a0," per A) The ratio of k=0, k=2 and k=4 transition strength is:",RjRdd(q)) -- define some transition operators qtheta=0 qphi=0 Tq001 = TnIXS_dd(q,qtheta,qphi) qtheta=Pi/2 qphi=Pi/4 Tq110 = TnIXS_dd(q,qtheta,qphi) qtheta=acos(sqrt(1/3)) qphi=Pi/4 Tq111 = TnIXS_dd(q,qtheta,qphi) qtheta=acos(sqrt(9/14)) qphi=acos(sqrt(1/5)) Tq123 = TnIXS_dd(q,qtheta,qphi) -- calculate the spectra nIXSSpectra = CreateSpectra(Hamiltonian, {Tq001, Tq110, Tq111, Tq123}, psiList, {{"Emin",-1}, {"Emax",6}, {"NE",3000}, {"Gamma",0.1}}) -- print the spectra to a file nIXSSpectra.Print({{"file","NiOnIXS_dd.dat"}}) -- a gnuplot script to make the plots gnuplotInput = [[ set autoscale set xtic auto set ytic auto set style line 1 lt 1 lw 1 lc rgb "#FF0000" set style line 2 lt 1 lw 1 lc rgb "#0000FF" set style line 3 lt 1 lw 1 lc rgb "#00C000" set style line 4 lt 1 lw 1 lc rgb "#800080" set style line 5 lt 1 lw 3 lc rgb "#000000" set xlabel "E (eV)" font "Times,12" set ylabel "Intensity (arb. units)" font "Times,12" set out 'NiOnIXS_dd.ps' set size 1.0, 0.3 set terminal postscript portrait enhanced color "Times" 12 set yrange [0:6.5] plot "NiO_Experiment/NIXS_dd_JSR_16_469_2009" using 1:($2*0.01) title 'experiment' with filledcurves y1=0 ls 5 fs transparent solid 0.5,\ "NiOnIXS_dd.dat" using 1:(-$15 -$17 -$19 +3.25) title 'q // 111' with lines ls 3,\ "NiOnIXS_dd.dat" using 1:(-$21 -$23 -$25 +2.50) title 'q // 123' with lines ls 4,\ "NiOnIXS_dd.dat" using 1:(-$9 -$11 -$13 +1.75) title 'q // 011' with lines ls 2,\ "NiOnIXS_dd.dat" using 1:(-$3 -$5 -$7 +1.00) title 'q // 001' with lines ls 1 ]] -- write the gnuplot script to a file file = io.open("NiOnIXS_dd.gnuplot", "w") file:write(gnuplotInput) file:close() -- call gnuplot to execute the script os.execute("gnuplot NiOnIXS_dd.gnuplot") -- transform to pdf and eps os.execute("ps2pdf NiOnIXS_dd.ps ; ps2eps NiOnIXS_dd.ps ; mv NiOnIXS_dd.eps temp.eps ; eps2eps temp.eps NiOnIXS_dd.eps ; rm temp.eps")
The spectrum produced:
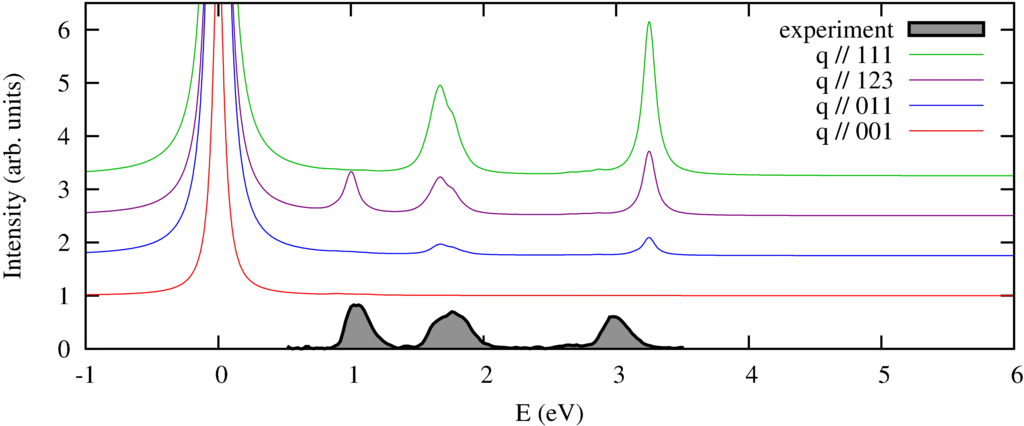 |
| nonresonant inelastic x-ray scattering spectra orientations of the momentum compared to the experimental spectra of a powder. |
|---|
We calculate the spectrum in 4 different directions of momentum transfer. The experimental spectra \cite{Verbeni:2009dx, Huotari:2008kx} are measured on a powder sample and thus do not show the strong momentum direction dependence. In previous \cite{Larson:2007ev} and subsequent \cite{Hiraoka:2009fa} this angular dependence has been observed.
For completeness the output of the script is:
- NIXS_dd.out
# <E> <S^2> <L^2> <J^2> <S_x^3d> <L_x^3d> <S_y^3d> <L_y^3d> <S_z^3d> <L_z^3d> <l.s> <F[2]> <F[4]> <Neg^3d> <Nt2g^3d><Neg^Ld> <Nt2g^Ld><N^3d> 1 -3.503 1.999 12.000 15.095 -0.370 -0.115 -0.370 -0.115 -0.741 -0.230 -0.305 -1.042 -0.924 2.186 5.990 3.825 6.000 8.175 2 -3.395 1.999 12.000 15.160 -0.002 -0.000 -0.002 -0.000 -0.003 -0.001 -0.322 -1.043 -0.925 2.189 5.988 3.823 6.000 8.178 3 -3.286 1.999 12.000 15.211 0.369 0.113 0.369 0.113 0.737 0.227 -0.336 -1.043 -0.925 2.193 5.987 3.820 6.000 8.180 for q= 4.5 per a0 ( 8.5037675605428 per A) The ratio of k=0, k=2 and k=4 transition strength is: 0.069703673179605 0.1609791731565 0.086144672158063