Density matrix plot
An intuitive way to look at eigenstates is to plot the charge density. For multi-electron wave-functions one can not plot the wave-function (it depends on $3\times n$ coordinates) but the charge density is a well defined simple quantity. We here use the rendering options of \textit{Mathematica} in combination with the package for \textit{Mathematica} (Quanty.nb) to plot the density matrix of the lowest three eigenstates in NiO.
The input script is:
- density_matrix_plots_Mathematica.Quanty
-- It is often nice to make plots of the resulting "wave-functions" Now one can not -- simply plot a multi-electron wavefunction \psi(r_1,r_2,r_3, ..., r_n) as it depends -- on 3n coordinates. -- One can plot the resulting charge density. -- This example calculates the density matrix of a state and then calls Mathematica to -- make a density plot of this matrix. -- the script for mathematica is included in the input file -- the input string needs calculated data which will be substituted before calling mathematica Verbosity(0) -- You need to change the absolute path in the script below mathematicaInput = [[ Needs["Quanty`PlotTools`"]; rho=%s; pl = Table[ Rasterize[ DensityMatrixPlot[IdentityMatrix[10] - rho[ [i] ], PlotRange -> {{-0.4, 0.4}, {-0.4, 0.4}, {-0.4, 0.4}}] ], {i, 1, Length[rho]}]; For[i = 1, i <= Length[pl], i++, Export["/Users/haverkort/Documents/Quanty/Example_and_Testing/History/current/Tutorials/20_NiO_Crystal_Field/Rho" <> ToString[i] <> ".png", pl[ [i] ] ]; ]; Quit[]; ]] NF=10 NB=0 dIndexDn={0,2,4,6,8} dIndexUp={1,3,5,7,9} OppSx =NewOperator("Sx" ,NF, dIndexUp, dIndexDn) OppSy =NewOperator("Sy" ,NF, dIndexUp, dIndexDn) OppSz =NewOperator("Sz" ,NF, dIndexUp, dIndexDn) OppSsqr =NewOperator("Ssqr" ,NF, dIndexUp, dIndexDn) OppSplus=NewOperator("Splus",NF, dIndexUp, dIndexDn) OppSmin =NewOperator("Smin" ,NF, dIndexUp, dIndexDn) OppLx =NewOperator("Lx" ,NF, dIndexUp, dIndexDn) OppLy =NewOperator("Ly" ,NF, dIndexUp, dIndexDn) OppLz =NewOperator("Lz" ,NF, dIndexUp, dIndexDn) OppLsqr =NewOperator("Lsqr" ,NF, dIndexUp, dIndexDn) OppLplus=NewOperator("Lplus",NF, dIndexUp, dIndexDn) OppLmin =NewOperator("Lmin" ,NF, dIndexUp, dIndexDn) OppJx =NewOperator("Jx" ,NF, dIndexUp, dIndexDn) OppJy =NewOperator("Jy" ,NF, dIndexUp, dIndexDn) OppJz =NewOperator("Jz" ,NF, dIndexUp, dIndexDn) OppJsqr =NewOperator("Jsqr" ,NF, dIndexUp, dIndexDn) OppJplus=NewOperator("Jplus",NF, dIndexUp, dIndexDn) OppJmin =NewOperator("Jmin" ,NF, dIndexUp, dIndexDn) Oppldots=NewOperator("ldots",NF, dIndexUp, dIndexDn) -- define the coulomb operator -- we here define the part depending on F0 seperately from the part depending on F2 -- when summing we can put in the numerical values of the slater integrals OppF0 =NewOperator("U", NF, dIndexUp, dIndexDn, {1,0,0}) OppF2 =NewOperator("U", NF, dIndexUp, dIndexDn, {0,1,0}) OppF4 =NewOperator("U", NF, dIndexUp, dIndexDn, {0,0,1}) -- Akm = {{k1,m1,Akm1},{k2,m2,Akm2}, ... } Akm = PotentialExpandedOnClm("Oh", 2, {0.6,-0.4}) OpptenDq = NewOperator("CF", NF, dIndexUp, dIndexDn, Akm) Akm = PotentialExpandedOnClm("Oh", 2, {1,0}) OppNeg = NewOperator("CF", NF, dIndexUp, dIndexDn, Akm) Akm = PotentialExpandedOnClm("Oh", 2, {0,1}) OppNt2g = NewOperator("CF", NF, dIndexUp, dIndexDn, Akm) Hamiltonian = 10.0 * OppF2 + 6.25 * OppF4 + 2.0 * OpptenDq + 0.06 * Oppldots + 0.000001 * (2*OppSz + OppLz) -- we now can create the lowest Npsi eigenstates: Npsi=3 -- in order to make sure we have a filling of 2 electrons we need to define some restrictions StartRestrictions = {NF, NB, {"1111111111",8,8}} psiList = Eigensystem(Hamiltonian, StartRestrictions, Npsi) oppList={Hamiltonian, OppSsqr, OppLsqr, OppJsqr, OppSz, OppLz, Oppldots, OppF2, OppF4, OppNeg, OppNt2g} print(" <E> <S^2> <L^2> <J^2> <S_z> <L_z> <l.s> <F[2]> <F[4]> <Neg> <Nt2g>"); for key,psi in pairs(psiList) do expvalue = psi * oppList * psi for k,v in pairs(expvalue) do io.write(string.format("%6.3f ",v)) end; io.write("\n") end io:flush() function tableToMathematica(t) local ret = "{ " for k,v in pairs(t) do if k~=1 then ret = ret.." , " end if (type(v) == "table") then ret = ret..tableToMathematica(v) else ret = ret..string.format("%18.15f + (%18.15f) I ",Complex.Re(v),Complex.Im(v)) end end ret = ret.." }" return ret end rhoList = DensityMatrix(psiList, {0,1,2,3,4,5,6,7,8,9}) rhoListMathematicaForm = tableToMathematica(rhoList) file = io.open("densitymatrix.nb", "w") file:write( mathematicaInput:format( rhoListMathematicaForm ) ) file:close() os.execute("/Applications/Mathematica08.app/Contents/MacOS/MathKernel -run '<<densitymatrix.nb'") os.execute("convert Rho1.png -transparent white temp.png ; convert temp.png Rho1.eps ; rm temp.png") os.execute("convert Rho2.png -transparent white temp.png ; convert temp.png Rho2.eps ; rm temp.png") os.execute("convert Rho3.png -transparent white temp.png ; convert temp.png Rho3.eps ; rm temp.png")
The produced figures are:
| $S_z=-1$ | $S_z=0$ | $S_z=1$ |
|---|---|---|
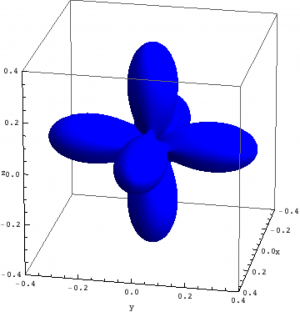 | 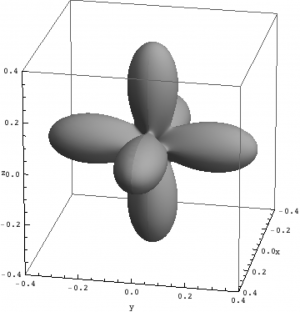 | 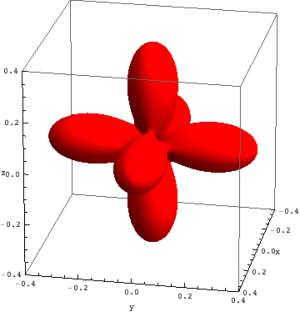 |
| Density matrix of the ground, first and second excited state of Ni in NiO. | ||
The plots can be seen in the figure above. We show the hole density (identity matrix minus the density matrix) which has lobes towards the O atoms. Ni in NiO has a hole in the $x^2-y^2$ and in the $z^2$ orbital. The color represent the spin down, $S_z=0$ and spin up state of the lowest triplet.
The output to the screen is:
- density_matrix_plots_Mathematica.out
<E> <S^2> <L^2> <J^2> <S_z> <L_z> <l.s> <F[2]> <F[4]> <Neg> <Nt2g> -18.093 2.000 12.000 14.471 -0.999 -0.119 -0.147 -1.020 -0.878 2.002 5.998 -18.093 2.000 12.000 14.471 -0.000 -0.000 -0.147 -1.020 -0.878 2.002 5.998 -18.093 2.000 12.000 14.471 0.999 0.119 -0.147 -1.020 -0.878 2.002 5.998 Mathematica 8.0 for Mac OS X x86 (64-bit) Copyright 1988-2011 Wolfram Research, Inc. Loaded the Quanty : Plot Tools Package - version 2016.4.13 Written by Maurits W. Haverkort